消化在线: 曲美他嗪通过调控MAPK-micro
来源:
曲美他嗪通过调控MAPK-microRNA通路抑制糖尿病性心肌病的心肌损伤
立题依据
2.1 研究意义
糖尿病是一种常见的代谢障碍疾病,并发症多,如动脉硬化、高血压等,严重威胁着人类生命。而糖尿病性心肌病(diabetic cardiomyopathy, DCM)是糖尿病患者的一种独立心脏并发症(不依赖心肌缺血和高血压),表现为左心室肥大、功能障碍,其特征为心脏舒张障碍先于收缩障碍,伴有心肌形态学和生物化学指标异常。目前,DCM的病理生理机制尚未明确,现有的DCM防治方法也不理想。因此,深入探讨DCM的发病机制及寻找新的防治策略、有效的药物和内源性信号分子,对防治DCM具有极其重要的理论意义与临床应用价值。
2 .2 国内外研究现状及发展动态
(1)p38MAPK通路在DCM的心肌炎症、纤维变性及氧化应激中的作用
DCM的发病机制比较复杂,涉及多种病理机制,如微血管病变、心肌纤维化(fibrosis)、间质炎症、氧化应激损伤及钙内稳态异常等[1]。其中心肌炎症被认为是DCM重要的病理生理机制之一,在链脲霉素(streptozotocin, STZ)诱导的DCM模型,Westermann等[2]研究证实,白细胞介素 (IL)-1β、IL-6、肿瘤坏死因子 (TNF)-α、核因子(NF)-κB和诱导型一氧化氮合酶(iNOS)等炎症因子明显增多;这些炎症因子继而导致心肌氧化应激(oxidative stress, OS)和心肌纤维化,在这些因素的共同影响下最后引起心室收缩和舒张功能障碍[3]。
Anderson等[7]报道,DCM心肌的活性氧(reactive oxygen species, ROS)生成明显增多,如心肌线粒体内H2O2水平升高[4]。ROS可通过氧化蛋白质、催化脂类物质为活性过氧化脂质及损伤DNA[5]等方式损伤线粒体及其他细胞成分,甚
至引起心肌细胞凋亡[5]。在2型糖尿病小鼠,由于心肌线粒体功能及ATP合成受损,从而导致心功能障碍及脂质堆积[6,7]。
重要的是,随着研究的进一步深入,学者们发现,丝裂原激活蛋白激酶(mitogen-avtivated protein hinase, MAPK)家族中的重要成员之一,p38MAPK在DCM的发生中被激活[2]。在哺乳类的多种实验模型,p38MAPK通路能被环境应激及炎症细胞因子所激活,并参与细胞死亡[8,9]。有报道指出,p38MAPK通路是糖尿病血管病变的共同炎症通路[10]。在STZ诱导的DCM模型,磷酸化(代表被激活)的p38MAPK表达明显增多[2]。具有抗炎及抗氧化作用的大麻二酚在抑制p38MAPK通路的同时,也能明显抑制炎症因子(如TNF-α、iNOS、 NFκ-B)及细胞凋亡[2],提示p38MAPK通路与DCM的炎症及细胞凋亡密切相关,通过p38MAPK通路引起的炎症可能在DCM的发生发展上起着重要的作用[11]。另一方面,通过抑制p38MAPK通路也能抑制ROS过度生成。因此,有学者[2]认为,DCM的特征之一是p38MAPK通路被激活,从而引起心脏炎症、纤维化及ROS生成增多。因此,寻找新型的既能抑制p38MAPK通路又能抑制炎症的药物,特别是体内又能产生的内源性信号分子对防治DCM具有重大的临床治疗价值。
(2)microRNA是一个阐明DCM发病机制的新思路
miR-375是胰岛细胞内最丰富的miRNAs, miR-375呈负调节葡萄糖引起的胰岛素(insuline)分泌[12]。抑制miR-375可增加胰岛素分泌,而过表达miR-375可通过减少肌侵蛋白(myotrophine, mtpn,是一种参与胰岛素颗粒融合的蛋白)表达继而损伤胰岛素的分泌[12,13]。另外,用前炎症细胞因子处理小鼠胰腺瘤细胞株(MIN6细胞)可引起miR-21,miR-34a和miR-146增多[14],阻断这些miRNAs可保护β细胞对抗这类炎症因子引起的死亡。MIN6细胞的miR-34a过表达能使抗凋亡蛋白Bcl-2表达减少[15],阻断miR-34a或miR-146可部分抑制软脂酸(palmitate)引起的β细胞凋亡[15]。
miR-133特异表达于心肌和骨骼肌,它调控心肌肥大,在心衰和心肌肥大时,miR-133表达下调[16]。糖尿病的心肌组织中miR-133表达也下调。此外,
高糖(high glucose)培养新生大鼠心肌细胞能引起miR-133表达下调[17],相反,转染miR-133可阻断高糖引起的心肌细胞肥大[18]。进一步研究还显示,miRNAs过度表达与DCM的心肌细胞凋亡及线粒体损伤密切相关[19,20]。Perry等[21]证实,miRNA-21表达增多与DCM的心肌纤维化有关。上述的研究结果表明,miRNA异常表达是糖尿病及其心肌细胞损伤(包括炎症)的病理机制之一,目前,探讨miRNA与DCM的关系才刚刚开始,但是,探讨miRNA差异表达在DCM发生发展中的作用,对于揭示其发病的基因机制,设计合理的治疗药物及判断预后,进一步提高DCM的治疗效果具有重要的意义,可能是一个新的突破口。
(3) 曲美他嗪具有心肌保护作用、抑制炎症、氧化应激、 MAPK通路等作用可为防治DCM提供新的策略
1) 曲美他嗪具有重要的心肌代谢保护作用
曲美他嗪[Trimetazidine,TMZ,化学名为1-(2,3,4-三甲氧苄基)呱嗪盐酸盐]是一种优化心肌能量代谢的抗心肌缺血药物。正常心肌的能量来源主要是葡萄糖和脂肪酸氧化代谢。前者分为无氧酵解和有氧氧化。TMZ的药理作用机制主要在于影响细胞尤其是心肌细胞的代谢。TMZ通过激活丙酮酸脱氢酶或者抑制内毒碱棕榈酰基转运酶-1,达到抑制脂肪酸的氧化磷酸化,使心肌细胞的氧化底物从脂肪酸转变为葡萄糖,促进葡萄糖的利用,提高心肌细胞产生能量的效率并由此减少脂肪酸氧化后带来的副作用[22]。Mody等[23]采用正电子扫描成像技术研究兔心肌组织葡萄糖的代谢率(rGMU)。他发现冠脉结扎前给予TMZ,无论是缺血区心肌组织还是缺血区周边心肌组织,rGMU均明显升高。有学者报道[24],经99mTc成像方法研究证明TMZ能维持缺血心肌的细胞代谢。由此可见,TMZ改善心肌代谢状况,使其在缺氧情况下,耗氧减少,产生能量的效率提高。
2) 曲美他嗪对心肌细胞线粒体的保护作用
线粒体在心肌细胞中起两方面的重要作用:(1)合成ATP;(2)维持Ca2+的平衡,二者都有赖于H+电化学梯度得以维持正常。在生理状况下,TMZ能提高Ca2+通透性,从而提高整个心肌细胞的能量代谢[25]。在缺氧状况下,线粒体内Ca2+超负荷可以引起线粒体肿胀、氧化磷酸化障碍。TMZ可与膜上的可通透性蛋白结合并使之失活以抑制Ca2+引起的线粒体肿胀[26]。Veitch等[27]研究了TMZ对呼
吸链的影响,他发现以抗坏血酸一丁二胺为基质时,TMZ能保护其缺氧状态下呼吸链的活性,通过MAPK/AKT通路抑制细胞凋亡。
3) 曲美他嗪具有提高组织缺氧耐受力的作用
心肌细胞膜和肌浆网的完整性对于维持细胞功能有重要意义。缺血及再灌注时都会造成其结构的破坏。Ruiz-meana等[28]发现培养的心肌细胞预先给予TMZ 100μmol/L,在缺氧状态下,心肌细胞的乳酸脱氢酶(LDH)渗出量明显低于对照组。台盼兰染色试验显示,TMZ组中细胞肌浆网完整性的比率显著高于对照组。Fantin等[29]亦有类似发现,并且认为它能够提高心肌对缺氧的耐受力是与其改变脂类代谢有关。Scntex等[30]发现TMZ能干预磷脂代谢,明显提高3-肌醇的利用率,从而提高细胞膜上磷脂的转换率,维持其缺氧状态下的膜结构稳定状态。
4) 曲美他嗪能消除氧自由基
氧自由基在细胞损伤中,特别是在心肌缺血和再灌注损伤中起重要作用,电子自旋共扼术可以直接检测超氧阴离子的存在。Maupoil等[31]发现,在离体大鼠心脏再灌注液中加入TMZ后,再灌注区氧自由基浓度下降20%。Guarniri等[32]通过酶学及化学荧光法,已证明了TMZ能对抗黄嘌呤过氧化酶催化氧自由基的产生,通过ROS/CTGF通路抑制心肌纤维化和炎症反应。
5) 曲美他嗪具有调节心肌细胞PH值的作用
细胞内pH值对于细胞代谢也具有重要影响,尤其是在缺氧状况下,细胞内酸化不断加深,细胞功能会严重受损。心肌缺血时,由于无氧糖酵解增强及血流冲洗作用停滞,酸性代谢产物在细胞内聚积,细胞内pH明显下降,当恢复血流灌注后心肌的细胞内pH可迅速恢复正常,同时却伴随着缺血心肌损伤的加重。这是由于再灌注恢复时,激活了Na+-H+交换,在排出细胞内H+的同时,使大量Na+进入细胞内,激活Na+-Ca2+交换,导致细胞内Ca2+超负荷,从而加重缺血心肌的损害。TMZ能调节心肌细胞内pH值,明显提高细胞内对酸的缓冲能力达55%~65%,减慢氯化铵带来的细胞内酸化[33]。TMZ的这种作用呈剂量依赖性。如果给予TMZ达2~3小时,则经Na+-H+交换被转入胞内的酸将显著降低。
6) 曲美他嗪在冠心病合并糖尿病方面的临床作用
糖尿病患者存在代谢紊乱,心肌的代谢由利用葡萄糖变为氧化游离脂肪酸,氧的利用效率减低,这样就降低了心脏机械效能。有研究证实早期糖尿病患者,
图1 曲美他嗪对心肌细胞保护的机制示意图。
1.曲美他嗪部分抑制了β脂肪酸的氧化,增加丙酮酸脱氢酶,增加葡萄糖氧化,有利于心功能不全的氧化利用。2.抑制钠和钙化的超载,以及酸中毒。3.减少氧化应激导致的细胞损伤,通过ROS/CTGF通路抑制心肌纤维化和炎症反应。4.通过MAPK/AKT通路抑制细胞凋亡,5.减少非偶联蛋白和增加PCr/ATP利用率。
即使没有明显心肌缺血表现,其运动耐量就已减低[34]。糖尿病患者由于代谢紊乱,更易损伤血管内皮功能。血管内皮功能异常伴发于心血管事件的全部过程, ET-1与心肌功能障碍的严重程度和预后相关。
传统的抗心肌缺血药物主要是通过的改变血流动力学,减少心肌的氧耗来治疗心绞痛,并不能提高缺血阈值。曲美他嗪则是通过提高氧利用率,保证心肌的氧化供能,提高了静息状态下心脏的机械效能,减轻了运动过程中心肌缺血的程度,最终提高了缺血阈值。在糖尿病患者心肌葡萄糖利用降低的情况下,改善葡萄糖代谢作用的曲美他嗪无疑适合患者的治疗。糖尿病本身就是心力衰竭独立的危险因素。在心力衰竭的各个时期,均有心肌能量代谢的损害。因为,心室重构使单位重量的心肌毛细血管数目减少,氧的弥散间距增大,故心肌缺氧。缺氧导致脂肪酸代谢增加,这就降低了心肌做功的机械效能。另外,有体外实验表明,衰竭心肌中ATP酶的活性约降低20% ~30%。ATP酶活性的降低使心肌能量利用发生障碍,因而心肌收缩性减弱。曲美他嗪可以使心肌耗能的优先底物从脂肪酸转向葡萄糖代谢,从而提高心肌的机械效率。通过核素显像技术,可以看到曲美他嗪通过改善心肌代谢,减少冬眠心肌的面积,改善左室功能[35]。
综上所述,可以确定曲美他嗪具有明显的抗炎症、抗氧化应激、线粒体保护作用及抑制MAPK 通路的作用,而且曲美他嗪可通过抑制MAPK 通路对抗多种伤害性刺激引起的细胞损伤、炎症反应、凋亡及氧化应激。鉴于MAPK 通路、炎症及氧化应激是导致糖尿病心肌病(DCM)的重要病理生理机制,虽然糖尿病伴缺血性心肌病有着明显疗效,但在DCM的治疗作用却不甚明确,因为前者是冠状动脉大血管的明显狭窄,而后者是冠状动脉微小血管的狭窄和损伤,其发病机制有着明显的差别。本实验的目的提出曲美他嗪治疗DCM的假说,并阐明曲美他嗪治疗DCM的细胞机制及在microRNA中的作用,也为进一步更好的应用临床提供更为详实的理论依据。
为探讨上述问题,本课题已开展了部分预实验,获得预期的结果。观察到高浓度葡萄糖可抑制H9c2 心肌细胞的存活率,促进活性氧(ROS)生成,增加凋亡细胞数量及降低线粒体膜电位(MMP)(上述预实验结果见研究基础部分)等,这为开展本课题研究打下了良好基础。
参考文献
[1] Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007; 115(25):3213-23.
[2] Westermann D, Walther T, Savvatis K, Escher F, Sobirey M, Riad A, Bader M, Schultheiss HP, Tsch?pe C. Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes, 2009 ;58(6):1373-1381.
[3] Westermann D, Rutschow S, J?ger S, Linderer A, Anker S, Riad A, Unger T, Schultheiss HP, Pauschinger M, Tsch?pe C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy:the role of angiotensin type 1 receptor antagonism. Diabetes, 2007, 56(3):641-6.
[4] Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart . J Am Coll Cardiol, 2009, 54(20):1891-8.
[5] Cimbaljevi? B, Vasilijevi? A, Cimbaljevi? S, Buzadzi? B, Kora? A, Petrovi? V, Jankovi? A, Kora? B. Interrelationship of antioxidative status, lipid peroxidation, and lipid profile in insulin-dependent and non-insulin-dependent diabetic patients. Can J Physiol Pharmacol, 2007;85(10):997-1003.
[6] How OJ, Aasum E, Kunnathu S, Severson DL, Myhre ES, Larsen TS. Influence of substrateupply on cardiac efficiency as measured by pressure-volume analysis ex vivo mouse hearts. Am J Physiol Heart Circ Physiol, 2005, 288(6):H2979-85.
[7] Tanaka Y, Konno N, Kako KJ. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc Res, 1992, 26(4):409-14.
[8] Lan A, Liao X, Mo L, Yang C, Yang Z, Wang X, Hu F, Chen P, Feng J, Zheng D, Xiao L. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLOS one,2011,6(10):e25921.
[9] Liu AL, Wang XW, Liu AH, Su XW, Jiang WJ, Qiu PX, Yan GM. JNK and p38 were involved in hypoxia and reoxygenation-induced apoptosis of cultured rat cerebellar granule neurons. Exp Toxicol Pathol. 2009;61(2):137-43.
[10] Murarka S, Movahed MR. Diabetic cardiomyopathy. J Card Fail, 2010; 6(12):971-979.
[11] Westermann D, Rutschow S, Van Linthout S, Linderer A, Bücker-G?rtner C, Sobirey M, Riad A, Pauschinger M, Schultheiss HP, Tsch?pe C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus.Diabetologia, 2006;49(10):2507-2513.
[12] Li Y, Xu X, Liang Y, Liu S, Xiao H, Li F, Cheng H, Fu Z. miR-375 enhances palmitate-induced lipoapoptosis in insulin-secreting NIT-1 cells by repressing myotrophin (V1) protein expression. Int J Clin Exp Pathol. 2010;3(3):254-64.
[13] Norlin S, Ahlgren U, Edlund H. Nuclear factor-{kappa}B activity in {beta}-cells is required for glucose-stimulated insulin secretion.Diabetes. 2005;54(1):125-32.
[14] Tang X, Muniappan L, Tang G, Ozcan S. Identification of glucose-regulated miRNAs from pancreatic {beta} cells reveals a role for miR-30d in insulin transcription. RNA. 2009;15(2):287-93.
[15] Roggli E, Britan A, Gattesco S, Lin-Marq N, Abderrahmani A, Meda P, Regazzi R. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells. Diabetes. 2010;59(4):978-86.
[16] Tian R, Abel ED. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation. 2001;103(24):2961-6.
[17] Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A.Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111(13):1652-9.
[18] Rossing P, Breum L, Major-Pedersen A, Sato A, Winding H, Pietersen A, Kastrup J, Parving HH. Prolonged QTc interval predicts mortality in patients with Type 1 diabetes mellitus. Diabet Med. 2001;18(3):199-205.
[19] Singh GB, Sharma R, Khullar M. Epigenetics and diabetic cardiomyopathy. Diabetes Res Clin Pract. 2011;94(1):14-21.
[20] Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res. 2012;93(4):583-93.
[21] Kumar A,Raut SK,Saikia UN,Sharma R,Khullar M. MicroRNA-21 contributes to diabetic cardiomyopathy.Circulation,2011;124:A15227.
[22] Stanley WC,Lopaschuk GD,Hall JL.Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions.Potential for pharmacological interventions.Cardiovasc Res,1997,33(2):243-257.
[23] Mody FV,Schelbert H,Coyle K,et al.Mechanism of action of a noval metabolically active antianginal agent (trimetazidine) delineated by PET. J Am Coll Cardiol,1996,27(suppl A9):132A.
[24] Ciavolella M,Greco C,Tavolaro R,et al.Acute oral trimetazidine administration increases resting technetium 99m sestamibi uptake in hibernating myocardium.J Nucl Cardiol,1998,5(2):128-133.
[25] Ferrari R. The role of mitochondria in ischemic heart disease.J Cardiovasc Pharmacol,1996,28(suppl 1):S1-5.
[26] Morin D,Elimadi A,Sapena R,et al.Evidence for the existence of [3H]trimetazidine binding sites involved in the regulation of the mitochondrial permeability transition pore.Br J Pharmacol,1998,123(7):1385-1394.
[27] Veitch K,Maisin L,Hue L.Trimetazidine effects on the damage to mitochondrial fractions caused by ischemia and reperfusion.Am J Cardiol,1995,76(6):25B-30B.
[28] Ruiz-Meana M,Garcia-Dorado D,Julia M,et al. Pre-treatment with trimetazidine increase sarcolemmal mechanical resistance in reoxygenated myocytes. Cardiovasc Res,1996,32(3):587-592.
[29] Fantini E,Demaison L,Sentex E,et al.Some biochemical aspects of the protective effect of trimetazidine on rat cardiomyocytes during hypoxia and reoxygenation. J Mol Cell Cardiol,1994,26:949-958.
[30] Sentex E,Sergiel JP,Lucien A,et al.Trimetazidine increases phospholipid turnover in ventricular myocyte. Mol Cell Biochem,1997,175(1-2):153-162.
[31] Maupoil V,Rochette L,Tabard A,et al.Evaluation of free radical formation during low-flow ischemia and reperfusion isolated rat heart.Cardiovas Drugs Ther,1990,4(suppl 4):791-795.
[22] Guarniri C,Muscari C.Antioxy-radical properties of trimetazidine. Res Comnmm Chem Pathol Pharmocol,1989,64(2):215-225.
[33] Layadic-Gossmanh D,Le Prigenf K,Feuvary D,et al.Effects of trimetazidine on pHi regulation in the rat isolated ventricular myocyte.Br J Pharmacol,1996,117(5),831-838.
[34] FangZY, Sharman J, Prins JBetal. Determinants ofexercise capacity in patients with type 2 diabetes. Diabetes Care, 2005; 28 (7): 1643~1648.
[35 FeolaM, BiggiA, FranciniAetal. Placebo of trimetazidine (99m) Tc Tetrofosmin myocardial SPECT and low dose dobutamine echocardiography in hibernatingmyocardium. ArchMed Res, 2006; 37 (1): 117~122.
1.3 拟解决的关键问题
目前国内外的临床和基础实验均已证实,曲美他嗪具有重要的心肌保护作用。在此研究基础上,本课题拟重点解决以下新的医学科学问题:
(1)曲美他嗪能否保护心肌细胞对抗DCM和高浓度Glu(模拟DCM)引起的心肌细胞凋亡、炎症反应、氧化应激、线粒体损伤及miRNA-133a表达的抑制作用;
(2)曲美他嗪是否通过抑制MAPK通路及上调miRNA-133a表达对抗DCM和高糖引起的心肌损伤。通过上述研究为防治DCM 提供新的治疗策略,为研究新型的抗DCM 药物提供科学的实验依据。
研究目标与研究内容
(1)明确miRNA(miR-133a和miR-21)及曲美他嗪在DCM发病机制中的作用
(2)明确MAPK通路是否通过抑制miR-133介导DCM的心肌损伤作用;
(3)明确曲美他嗪能否通过抗凋亡、抗炎症、抗氧化、线粒体保护、上调miR-133a表达等作用及机制保护心肌细胞对抗DCM或高糖引起的损伤;
(4)明确曲美他嗪是否通过抑制MAPK通路保护心肌细胞对抗DCM或高糖引起的损伤;
通过上述目标,为进一步阐明DCM的发病机制及曲美他嗪抑制DCM作用与机制提供新颖的实验资料,为防治DCM开拓新思路,为研发新型的防治DCM药物提供实验依据。
3.2 研究内容
本课题拟在糖尿病性心肌病(DCM)大鼠模型(在体)及高糖损伤心肌细胞模型(离体)分别从基因水平和分子水平探讨DCM的发病机制及曲美他嗪抑制DCM心肌细胞损伤(包括细胞凋亡、炎症因子分泌、线粒体受损、氧化应激、microRNA表达改变等)及其作用机制,特别是抑制MAPK通路在其中的作用。
(1)在DCM大鼠模型开展的研究内容(与正常大鼠比较)
①观察心肌细胞miR-133a和miR-21表达水平的改变;
②观察心肌细胞凋亡情况(观察指标:凋亡细胞百分比、凋亡相关蛋白caspase-3和caspase-9表达水平);
③观察心肌细胞的炎症反应(观察炎症因子包括:白细胞介素-1β(IL-1β)、IL-6、IL-8、NF-κB、TNF-α等)的情况;
④了解心肌细胞的氧化应激反应(测定ROS水平和还原型谷胱甘肽(GSH)的水平)
⑤探讨心肌细胞的线粒体损伤(观察指标MMP和细胞色素C表达水平);
⑥观察心肌细胞磷酸化(p)MAPK蛋白表达水平;
⑦观察曲美他嗪对DCM心肌细胞凋亡的抑制作用(指标同上);
⑧观察曲美他嗪对DCM心肌细胞miR-133a和miR-21表达的作用;
⑨明确曲美他嗪对DCM心肌细胞炎症损伤抑制作用(指标同上);
⑩探讨曲美他嗪对DCM心肌细胞ROS水平和GSH水平的作用;
?了解曲美他嗪对DCM心肌细胞线粒体的保护作用(指标同上);
?明确曲美他嗪对DCM心肌细胞p-MAPK表达的抑制作用;
?了解MAPK通路是否参与DCM心肌细胞凋亡、炎症、线粒体损伤、氧化应激及miRNAs(miR-133a和miR-21)表达改变;
(2)在高糖(30 mmol/L)损伤H9C2心肌细胞模型(离体实验)开展的研究:
具体研究内容如下:
?建立高浓度葡萄糖(Glu)损伤心肌细胞(H9c2 细胞)模拟DCM 的实验模型,观察高浓度Glu对下列指标的影响:
A.炎症因子:白细胞介素IL-1β (IL-1β)、IL-6、IL-8、 NFκ-B 等;
B.心肌细胞凋亡指标:细胞凋亡数量、caspase-3 表达、caspase-9 表达;
C.线粒体损伤指标:线粒体膜电位(MMP)、细胞色素C表达水平;
D.曲美他嗪合成酶(CES)表达及活性;
E.氧化应激:活性氧(ROS)水平,还原型谷胱甘肽(GSH);
F.磷酸化(p)MAPK 表达;
G.通过以上实验明确高浓度Glu 对心肌细胞的致凋亡作用、线粒体损伤、致炎症作用和氧化应激及机制,以检验实验模型模拟DCM 的可靠性。
?明确曲美他嗪能否抑制高浓度Glu 引起的心肌细胞炎症反应,观察指标同上述⑴的炎症因子项。
?明确曲美他嗪能否抑制高浓度Glu 引起的心肌氧化应激反应,观察指标包括活性氧水平,还原型谷胱甘肽水平;
?明确曲美他嗪能否保护心肌细胞对抗高浓度的Glu 引起的细胞凋亡,观察指标为上述的①B的心肌细胞凋亡指标;
?明确曲美他嗪能否抑制高浓度Glu 对心肌细胞MAPK 通路的激活,观察指标为磷酸化MAPK表达水平;
?明确曲美他嗪能否对抗高糖对心肌细胞miRNA-133a和miRNA-21的调控作用;
?应用抗miRNA-133a抑制剂转染(anti-miRNA-133a inhibitor transfection)技术观察能否阻断曲美他嗪的心肌保护作用(包括抗炎、抗凋亡、抗氧化等);
明确MAPK 通路在高浓度Glu 损伤心肌细胞及在曲美他嗪 心肌细胞保护中的作用:
a.高浓度Glu 处理心肌细胞前30 min 应用特异性MAPK 抑制剂SB203580 处理心肌细胞,观察SB203580 能否抑制炎症因子(同上)、氧化应激(ROS、GSH)、细胞损伤(细胞存活率、细胞凋亡、MMP、Caspase-3和Caspase-9 表达)及改善CSE 表达水平及活性;
b.应用RNA 干扰技术沉默心肌细胞的MAPK 后观察对①项各指标的影响。
miRNAs:miRNA-133a和miRNA-21表达水平;
研究方案
4.1 研究方案及路线
(1)通过建立大鼠DCM模型,从下述6个层面
① 心肌细胞microRNA (miRNA-133a和miRNA-21)表达改变;②心肌细胞炎症;③心肌细胞凋亡;④心肌细胞氧化应激和线粒体损伤;⑤p38MAPK通路,分别阐明大鼠DCM的病理生理机制及曲美他嗪抑制糖尿病心肌病的作用与机制。
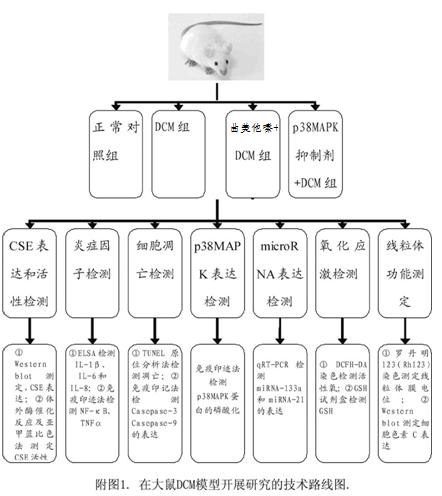
注:1. 建立大鼠DCM模型: 参照文献(董世芬,等.中国实验动物学报,2009)介绍的方法,高糖高脂饲养大鼠6周,在第6周一次性腹腔注射链脲佐菌素(FTZ 30mg/kg);注射后72 h,测定空腹血糖,如血糖水平连续2次高于11.1mmol/L视为构建糖尿病模型成功。随后,继续用高糖高脂饲料喂养“糖尿病”大鼠6周,根据文献证实,此时大鼠已患上糖尿病心肌病。在上述12周实验结束时行大鼠心肌细胞的分离,方法如下:打开Langendorff灌流系统,水温箱调节温度为37°C。用1000mL三蒸水反复冲洗Langendorff灌流系统后,加入200mL的无钙Tyrode’s液。选取体重150-200g的雄性Wistar大鼠。用3%的戊巴比妥钠腹腔内注射麻醉(40–50 mg/kg),然后腹腔注射200IU的肝素。开胸后迅速取出心脏,放入盛有预冷的无钙Tyrode’s液玻璃皿中,分离出主动脉,并剥离掉附属组织。 将主动脉挂于Langendorff灌流系统的针头上并系紧(主动脉插入深度2-4mm),用蠕动泵调节流速为15mL/min。5min后,换用80ml无钙Tyrode’s液含有32mg II型胶原酶,80mg 牛血清白蛋白,消化30min左右。将心脏剪下放入无钙Tyrode’s液中,用眼科剪剪碎成1mm2大小,用吸管吹打。37°C振荡10min以进一步释放细胞。200目滤网过滤后,用无钙Tyrode’s液定容至10mL。加入10uL 100mM CaCL2,室温静置10min。加入40uL 100mM CaCL2,室温静置10min。加入50uL 100mM CaCL2,室温静置10min, 最终得到的是心肌细胞。
(2)实验分组
雄性Wistar-kyoto鼠72只,随机分为6组,每组12只:① 正常对照组:用基础饲料喂养;② 糖尿病心肌病(DCM)组; ③ 曲美他嗪 (10mg﹒Kg-1﹒day-1) + DCM组:在注射STZ 72 h后,每天早上8 时ip注入曲美他嗪;④MAPK抑制剂+DCM组:在注射STZ 72h后,每天早上8点ip注入MAPK抑制剂SB203580 (15mg﹒Kg-1);⑤SiMAPK+高糖组;⑥抗miR-133a抑制剂+高糖组。[参考文献:常建辉,等,药学学报,2011]。正常对照组和DCM组则ip注射等容积的对照溶液(DMSO)。
(3)建立高糖损伤H9c2心肌细胞模型,从下述7个层面:
①心肌细胞miRNAs(miRNA-133a和miRNA-21)表达改变;②心肌细胞炎症;③心肌细胞凋亡;④心肌细胞线粒体损伤;⑤心肌细胞氧化应激反应;⑥MAPK通路,分别阐明高糖对心肌细胞损伤的病理生理机制及曲美他嗪对抗高糖对心肌细胞的损伤作用及机制。
注:1)按Lan AP,等人介绍的方法转染MAPK小干扰RNA(Si-p38MAPK)入心肌细胞[参考文献:Lan A, et.al. PLOS one,2011,6(10):e25921.]以沉默心肌细胞的p38MAPK基因。
2) 按Liu J 等人介绍的方法转染抗miR-133a抑制剂入心肌细胞[参考文献:Liu J,et al.Biochemical and Biophysical Research Communications.2011,413:342-347]
3)应用30mM葡萄糖损伤H9c2心肌细胞36 h。
4)应用20 μmol/L MAPK 特异性抑制剂SB203580预处理心肌细胞60 min, 再用30mM葡萄糖损伤心肌细胞36 h(即MAPK 特异性抑制剂+高糖组),以明确MAPK通路在高糖损伤心肌细胞中的作用。
5)观察不同浓度(100、200、300、400、500 μmol/l)曲美他嗪预处理心肌细胞 90 min对抗高糖诱导的心肌细胞凋亡作用(以PI染色流式细胞仪检测凋亡细胞百分比为观察指标),探讨是否存在浓度依赖关系,并根据实验结果筛选出最有效的保护浓度,例如400umol/L完成后续的实验。
6)明确以“有效的” 曲美他嗪浓度预处理心肌细胞不同时间(如20min、 30 min、60 min、 90 min、 120 min)后,是否存在抗细胞凋亡的时效依赖关系,从中筛选出最有效的预处理时间(假设90 min),并以此时间作用作为后续实验的预处理时间。
7) 观察“最有效的” 曲美他嗪浓度处理心肌细胞不同时间(20min、 30 min、60 min、 90 min、 120 min)对高糖上调p-MAPK表达的影响,明确曲美他嗪对MAPK通路的调控是否存在时效性。
4.2 可行性分析
(1)理论上可行
目前,MAPK 通路在糖尿病心肌病(DCM)的炎症、氧化应激及心肌纤维化发生中的作用日益受到重视,这一发病机制有可能成为防治DCM 的作用靶点。近年的研究表明曲美他嗪具有细胞保护作用,包括心肌保护、神经保护及皮肤细胞保护,其细胞保护机制可能与抗氧化、抗凋亡、抗炎症、抑制MAPK 通路达等有关。尽管国内外尚未见有关曲美他嗪是否对DCM 有作用的报道,但是,基于现有研究成果,我们提出“曲美他嗪能保护心肌细胞对抗DCM 损伤”这一工作假说具有充分的理论基础,符合逻辑推断。
(2)有一定的前期工作基础:
本申请课题是申请者近来对DCM心肌细胞保护作用及其作用机制研究的延续与扩展,业已证实曲美他嗪具有明显的细胞保护作用,包括心肌保护作用,此作用与其抑制活性氧(ROS)生成,促进还原型谷胱甘肽生成,抑制细胞毒性和细胞凋亡,保护线粒体,抑制炎症因子(包括IL-6、IL-8、NF-κB、iNOS/NO、COX-2 等),抑制MAPK 等有关。因此,这些前期研究为开展本课题研究提供扎实的研究基础。
(3)有技术力量保证:
申请者为医学博士,主治医师。长期从事心血管疾病的临床与科研工作。近年来采取临床与基础研究相结合的方式从事心肌损伤细胞保护作用及其作用机制的研究,获得一些研究成果,近5年来发表研究论文4篇,其中以第一作者发表SCI 收录期刊科研论文1篇(参考“研究条件与基础”部分),其他3篇,先后圆满地完成了1 项广东省科技计划项目及参与2项国家自然科学基金的研究工作。课题组人员组成合理,既有熟悉临床医学工作的医生和从事基础心血管研究的专家,又有博士研究生和硕士研究生。课题组成员熟悉和了解与本课题相关的国内外研究动态,掌握本研究需要用到的各种研究技术与方法,这从技术力量上保证本课题能够顺利完成。
(4)有实验条件保障:
申请者所在的实验室配备有完成本项目所需的各种仪器设备,可为完成相关的研究内容提供良好的技术支撑;实验所需的试剂或耗材等在国内外均有销售。
龙明